康復理療產品服務商-深圳孚瑞泰科技有限公司
語言選擇:


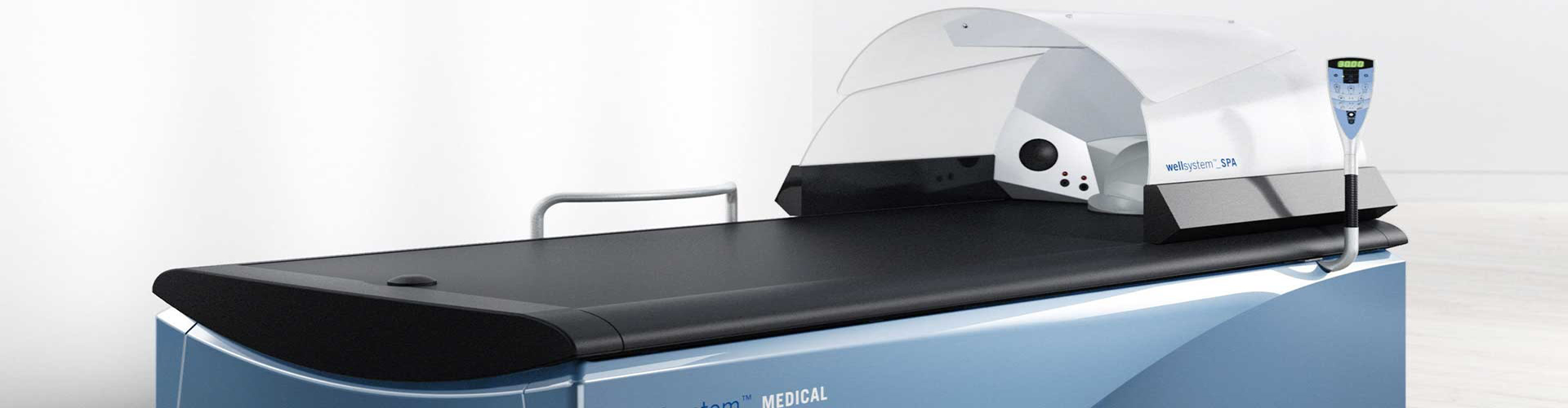

醫療器械指令93/42/EC(Medical Devices Directive93/42/EC amended by Directive 2007/47/EC)
各成員國有關醫療器械的安全、健康保護和工作特性的法律、法規和行政條款的內容和范圍是不同,各成員國之間對這類器械的認證和檢驗程序存在差異,這種差異在歐洲共同體內形成貿易壁壘;鑒于各國針對醫療器械的使用所制定的有關患者、使用者及其它人員的安全和健康保護的條款應予以協調,以保證此類器械在歐洲共同體內部市場自由流通;因而只要遵守歐洲共同體法律,這些條款并不影響各成員國實施上述措施的能力;因此保持和提高各成員國已達到的保護水平是本指令的基本目的之一
醫療器械MDD指令適用范圍很廣,包括除有源植入性和體外診斷器械之外的幾乎所有的醫療器械,如無源性醫療器械(敷料、一次性使用產品、接觸鏡、血袋、導管等);以及有源性醫療器械,如核磁共振儀、超聲診斷和治療儀、輸液泵等。該指令已于1995年1月1日生效,過渡截止日期為1998年6月13日從1998年6月14日起強制執行。指令規定,在指令正式實施后,只有帶有CE標志的醫療器械產品才能在歐盟市場上銷售。產品取得了CE認證,就可以帖上CE標志,但只限于申請認證時提交的TCF中所描述的產品,貼上CE標志意味著該產品可以進入歐盟市場和要求產品取得CE認證才能進入該地區的國家和地區。產品CE認證的有效期為五年。我們這次獲得的CE證書因所提交的TCF文件是此次產品認證的資料,因此CE標志只能加貼此產品。如產品有重大更改,必須向CE認證機構提交更改的文件資料重新認證,否則不能加貼CE標志。
鑒于實質上在合格評定程序中,必須將器械分為四個產品類別;鑒于分類原則的依據是有關器械技術設計和制造的潛在危險對人身的傷害程度;鑒于在一般情況下,由于第Ⅰ類器械傷害性較低,其合格評定程序可由制造商單獨完成,也可由歐盟指定機構完成;鑒于對第Ⅱa類器械,指定機構應在制造階段強制性介入;鑒于第Ⅱb類和第Ⅲ類器械有較高的潛在危險性,需要指定機構對器械的設計與制造進行檢驗;鑒于第Ⅲ類器械屬于最具危險的器械,它們在投放市場前需預先對其合格性作明確的核準
Class I低風險(Low risk)
Class IIa低到中風險(Low to medium risk)
Class IIb中風險(Medium risk)
Class III高風險(High risk)
醫療器械CE認證對以上幾個等級認證有共同的基本要求
基本要求是MDD的最重要部分:
1、器械設計和生產必須保證:按照其預定和條件使用,器械不會損害醫療環境、患者安全、操作者或其他人員的安全和健康;使用時的潛在危險與患者受益相比較可以為人們所接受,但應具有高水平的防護辦法。
2、生產者的設計和制造方案,必須考慮在現有工藝技術條件下遵守安全準則、生產者應:
首先,應盡可能降低甚至避免危險;
其次,對無法避免的危險采取適當的防護措施,包括安裝報警裝置;
最后,告知用戶所提供防護措施的弱點及其可能帶來的危險。
3、器械必須取得生產者期望獲得的功能。器械設計制造和包裝應有利于第一條(2)(A)D多規定的各項功能的發揮。
4、在生產線者確定的器械使用壽命期內,在正常使用可能出現的壓力,第1、2、3款所指的各項性能應保持穩定,不能危害醫療環境、危害患者、使用者或其他人員的健康。
5、器械的設計、生產和包裝應當保證,器械的性能在運輸和儲存過程中只要遵守有關規定不會發生根本逆變。
6、副作用的大小同器械的使用性能相比較可以為人們所接受。
7、化學、物理和生物性能。
8、感染和微生物污染。
9、組裝和環境因素。
10、檢測器械。
11、輻射防護。
12、帶有能源或與其他能源相連接的器械。
13、生產者提供的操作信息。
14、如果需要根據醫療數據確定器械是否滿足基本要求,如第六款的情形,有關數據必須按照附錄Ⅹ的規定取得。
聯系人: 邵小姐
手機:
電話:18088889812
郵箱:forelax@foxmail.com
地址:深圳市南山區
